在全球医疗器械监管体系中,日本凭借严谨的监管架构、规范的注册流程、完善的质量管控及专业化的执行机构,构建了覆盖医疗器械“上市前注册-上市后监督”全生命周期的监管体系。日本医疗器械监管以厚生劳动省(Ministry of Health, Labor and Welfare,MHLW)为统筹核心,其管辖的独立行政法人药品和医疗器械综合机构(Pharmaceuticals and Medical Devices Agency,PMDA)承担具体技术审评、审核及市场监督等职能。本文系统梳理日本医疗器械核心监管机构(PMDA与MHLW)的职能分工、全流程注册要求(重点适配外国制造商)、质量管理体系规范及上市后监督要求,深入解析各环节核心要点,为医疗器械企业(尤其是外国企业)拓展日本市场、合规开展注册申报及运营提供、、系统的行业参考(本文所指医疗器械不含体外诊断试剂)。
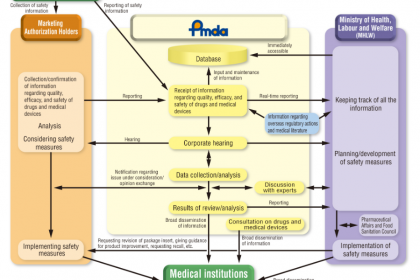
一、核心监管机构解析:PMDA与MHLW的职能分工与协同机制
日本医疗器械监管体系以“MHLW统筹管控、PMDA专业执行”为核心架构,两者权责清晰、协同联动,同时明确各类参与主体的监管要求,共同保障医疗器械的、有效与质量可控,为医疗器械注册与运营提供坚实的监管支撑。
(一)厚生劳动省(MHLW):统筹全局的主管部门
MHLW作为日本医疗器械监管的主管部门,核心使命是保护日本民众免受因药品和医疗器械不、无效或质量不达标造成的健康危害,聚焦宏观统筹、法规制定与最终审批,具体职能包括:
- 制造商注册管理:负责统筹医疗器械制造商(含日本本土及外国制造商)的注册审批与管理,规范制造场所合规性;
- 市场参与者许可:对医疗器械上市许可持有人(MAH)、经销商、维修服务提供商等核心参与者进行许可管理,明确准入标准;
- 法规与标准制定:颁布医疗器械相关部级法令、指导方针及行业标准,明确监管实施细则与技术要求(如第169号部令MO 169);
- 最终授权发布:作为医疗器械上市授权的最终决策部门,审定PMDA提交的审评结果,发布医疗器械上市授权;
- 机构监督管理:对PMDA的工作进行监督与指导,确保其技术审评、审核及市场监督工作合规、开展。
(二)药品和医疗器械综合机构(PMDA):专业执行的核心技术机构
PMDA全称为Pharmaceuticals and Medical Devices Agency,是MHLW管辖下的独立行政法人,成立于2004年,专注于医疗器械、药品、再生医学产品等的具体监管执行工作,是日本医疗器械注册与监管的核心技术支撑机构,核心职能包括:
- 授权程序执行:负责医疗器械上市授权的具体技术审评工作,开展监管审查,为MHLW的最终授权提供专业技术支撑;
- 专业咨询服务:就医疗器械目标临床研究方案、上市授权申报等事宜,为企业提供专业咨询与指导,助力企业合规申报;
- GMP合规检查:对医疗器械制造商(含本土及外国)开展良好生产规范(GMP)检查,核查制造场所与生产过程的合规性;
- 市场监督管控:承担医疗器械上市后市场监督工作,跟踪产品使用,处置不良事件与质量隐患;
- 质量管理体系审核:负责高风险医疗器械及部分中等风险医疗器械的质量管理体系(QMS)审核,确保企业质量管控达标。
(三)PMDA与MHLW的协同机制
PMDA隶属于MHLW,接受MHLW的监督与指导,两者形成“宏观统筹+专业执行”的协同模式:PMDA负责所有医疗器械的技术审评、GMP检查、QMS审核、市场监督等具体专业工作,提出审评意见与审核结论;MHLW基于PMDA的专业意见,负责法规制定、参与者许可、制造商注册统筹、最终授权发布等宏观管控工作,确保监管工作的专业性、性与统一性。
二、日本医疗器械全流程注册指南(重点适配外国制造商)
日本医疗器械注册流程规范、环节清晰,核心遵循“质量管理体系搭建—指定授权代表—制造商注册—产品分类—授权程序选择—技术文档提交—QMS审核—上市授权—上市后维护”的逻辑,其中外国制造商与日本本土制造商的注册要求存在差异,重点环节如下(共8个核心步骤):
Step 01:建立符合要求的质量管理体系(QMS)
作为外国制造商,无需提供“本国批准”证明即可申请日本医疗器械上市授权,但必须满足日本质量管理体系(J-QMS)要求,核心遵循2014年修订的第169号部令(MO 169)。
关键要点:
- MO 169与ISO 13485:2003基本一致,企业持有的ISO 13485证书可作为满足J-QMS要求的重要参考,降低审核难度;
- MO 169第3章包含额外要求,需企业(制造商)与上市许可持有人(MAH)共同遵守,包括文件保留期限、警戒程序要求,以及制造商与MAH之间的沟通规则;
- 质量管理体系需覆盖产品设计、生产、检验、仓储、销售等全流程,确保产品质量可控。
Step 02:指定日本当地授权代表(MAH)
外国制造商在日本申请医疗器械上市授权前,必须指定一名日本当地的上市许可持有人(Marketing Authorized Holder,MAH),MAH是外国制造商进入日本市场的核心衔接主体,对医疗器械在日本的合规运营承担全部责任,同时也是上市授权的所有者。
关键要点:
- MAH核心职责:不仅需位于日本、提交上市授权文件,还需对QMS承担全部责任,负责医疗器械及批次放行、上市后市场监督、不良事件报告等工作;
- MAH资质要求:并非所有实体均可担任MAH,需先向MHLW申请营业执照(称为KYOKA),执照分为三类,对应不同的医疗器械类别授权范围:
1. 类:可担任所有类别(Ⅰ、Ⅱ、Ⅲ、Ⅳ类)医疗器械的MAH;
2. 第二类:可担任Ⅰ类、Ⅱ类医疗器械的MAH;
3. 第三类:仅可担任Ⅰ类医疗器械的MAH。
Step 03:完成外国制造商注册(FMR)
在医疗器械获得日本上市授权前,外国制造商需在MAH的协助下,向MHLW完成外国制造商注册(Foreign Manufacturer Registration,FMR),所有负责产品开发、最终组装或生产的制造场所均需注册,组件制造商无需注册。
关键要点:
- 注册提交:通过Form 63-5提交注册申请,需提供完整、准确的相关信息,包括制造商名称及地址、所有拟上市设备清单、经理声明、合规负责人信息、生产现场详细信息等;
- 注册周期:属于行政审核程序,流程相对简便,从提交申请到完成注册约需30天;
- 注册有效期与维护:注册有效期为5年,到期后需及时更新;若制造商地址、联系人等信息发生变更,需在30天内通知MHLW。
Step 04:对医疗器械进行分类
产品分类是日本医疗器械注册的核心前提,需结合日本医疗器械术语集(Japanese Medical Device Nomenclature, JMDN)编码及产品风险等级,明确产品所属类别(Ⅰ类、Ⅱ类、Ⅲ类、Ⅳ类),不同类别对应不同的授权程序、审核机构与监管要求,具体分类标准如下:
- Ⅰ类(一般医疗器械):风险,如手术刀,监管方式为地方政府备案,无实质性审查;
- Ⅱ类(管制医疗器械):风险中等,如电子内窥镜,需通过第三方认证机构(RCB)审核;
- Ⅲ类、Ⅳ类(高度管制医疗器械):风险较高或,如透析器、起搏器,需通过PMDA技术审评,由MHLW发布最终授权。
Step 05:选择适配的上市授权程序
根据产品分类及特性,选择对应的上市授权程序,核心分为两类:
- 低风险产品(Ⅰ类):采用备案程序,完成地方政府备案后即可上市;
- 中高风险产品(Ⅱ、Ⅲ、Ⅳ类):采用审核/审评程序,Ⅱ类由RCB审核,Ⅲ、Ⅳ类由PMDA审评,审核/审评通过后由MHLW发布授权。
Step 06:编制并提交日语技术文档
技术文档是医疗器械上市授权审核/审评的核心依据,所有提交的文档必须为日语,提交格式遵循IMDRF国际公认的STED格式,核心包含STED概要文件及以下8个附件:
1. 附件A:开发记录(含先前设备版本、全球授权情况);
2. 附件B:产品规格(详细技术参数、性能指标等);
3. 附件C:稳定性和保质期数据(验证产品有效期);
4. 附件D:符合适用标准和基本原则的证明;
5. 附件E:性能测试数据(验证产品性能达标);
6. 附件F:风险分析报告(识别产品潜在风险及防控措施);
7. 附件G:制造相关资料(生产过程、质量监督、工艺等);
8. 附件H:临床数据(根据产品风险等级,提供相应的临床试验或临床验证数据)。
补充说明:文档审核过程中,企业可与监管机构(PMDA/RCB)进行公开沟通,若监管机构有疑问,文档审核预定持续时间不中断,确保审核推进。
Step 07:申请质量管理体系(QMS)审核
医疗器械上市授权审核/审评通过前,需完成QMS审核,审核机构根据授权程序确定(PMDA或RCB),审核由MAH提交申请,覆盖MAH的QMS及外国制造商注册生产场所的QMS,对每个医疗器械系列执行独立审核。
关键要点:
- 审核形式:分为现场检查与文件审核,具体采用哪种形式,由监管机构根据企业ISO 13485认证情况、先前审核结果、不良事件报告、制造过程复杂性等因素确定;
- 文件审核核心内容:包括制造现场概览、组织图、质量管理手册、质量管理文件清单、制造过程及验证资料、MAH合同、警戒程序等;
- 特殊说明:日本自2015年起加入医疗器械单一审计计划(MDSAP),接受MDSAP审计报告,通过MDSAP审计的制造商可避免重复审核,简化审核流程。
Step 08:维护上市监管,确保持续合规
医疗器械获得上市授权后,需进入常态化上市后维护阶段,由MAH牵头负责,严格遵循上市后监督要求,确保产品持续合规、有效,具体要求详见本章第三节(上市后监督)。
三、日本医疗器械上市后监督要求
日本医疗器械上市后监督以MAH为核心责任主体,核心目的是跟踪产品使用,及时处置不良事件与质量隐患,保障日本民众健康权益,相关要求明确规定于“良好警戒惯例”条例(部长条例135)中,核心要点如下:
- 不良事件报告责任:MAH有义务及时收集、整理医疗器械在日本市场使用过程中发生的不良事件,向制造商及监管机构(PMDA/MHLW)提交报告,不得迟报、漏报、瞒报;
- 警戒程序执行:MAH需建立完善的警戒程序,明确不良事件收集、评估、报告的流程与责任分工,确保快速响应、有效处置;
- 产品质量跟踪:MAH需对上市医疗器械的质量进行持续跟踪,定期开展质量回顾,及时发现并解决产品质量问题,必要时启动召回程序;
- 资料留存与更新:MAH需按要求留存医疗器械上市后相关资料(如不良事件报告、质量回顾报告等),确保资料可追溯;同时及时更新制造商注册信息、MAH信息等,确保合规性;
- 监管配合义务:MAH需积极配合PMDA/MHLW开展的上市后监督检查,提供相关资料与数据,不得拒绝、阻碍监督检查工作。
四、核心总结
日本医疗器械监管体系以“、有效、质量可控”为核心,形成了“MHLW统筹、PMDA执行、MAH牵头、全流程管控”的完善生态。MHLW负责宏观法规与最终授权,PMDA承担专业技术审评与审核,MAH作为外国制造商的核心衔接主体,承担着合规运营的主要责任。
对于外国医疗器械企业而言,想要顺利进入日本市场,需重点把握三大核心:一是搭建符合MO 169要求的质量管理体系,充分利用ISO 13485与MDSAP的优势简化审核流程;二是选择具备对应资质的MAH,规范完成外国制造商注册与产品分类;三是严格遵循全流程注册要求,规范提交技术文档,同时重视上市后监督,确保持续合规。
本文系统梳理的监管机构职能、注册流程、QMS要求及上市后监督规范,可为相关企业与从业者提供的参考,助力其完成合规申报与运营,把握日本医疗器械市场的准入要点与监管趋势。